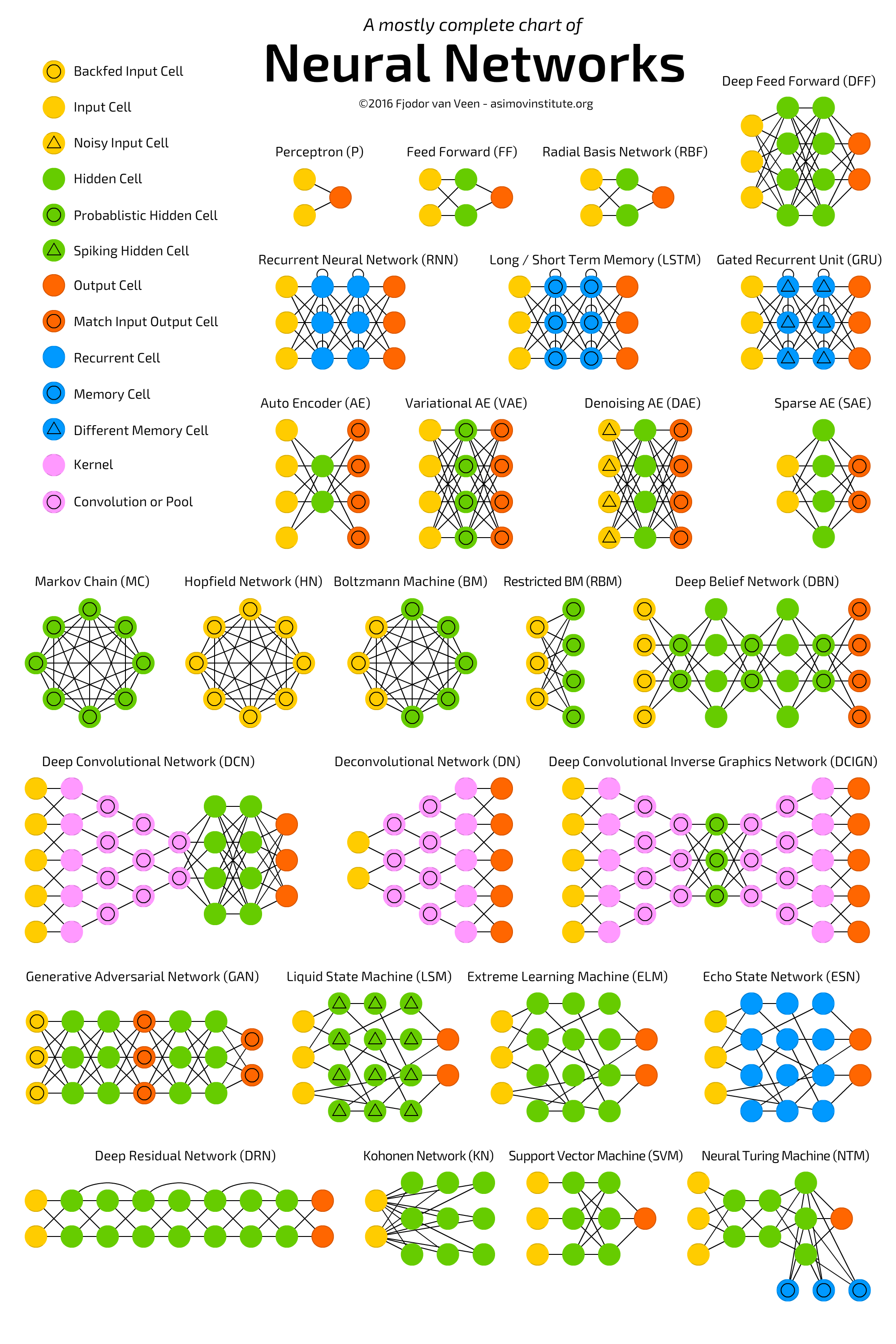
Programmierung für OttoNormalVerbraucher
Facebook und Co. arbeiten daran Nachrichten so aufzubereiten, dass sie emotional noch mehr ansprechen, als ob die gesellschaftliche Situation nicht schon aufgeheizt genug ist. Wir arbeiten daran dem Endnutzer Werkzeuge bereitzustellen um seine rationale Urteilskraft mit Hilfe des Computers zu stärken. Dafür benötigt man möglichst einfache aber dennoch leistungsstarke Programmiersprachen und umfangreiche, vertrauenswürdige, öffentlich zugängliche Informationen in Form von vielgestaltigen großen Tabellen und Dokumenten ähnlich der Wikipedia.
Auch wenn die entwickelte Sprache so einfach wie möglich ist, wird sie im Gegensatz zum Facebookansatz einen gewissen Lernaufwand erfordern.
Eine solche Programmiersprache in Kombination mit vertrauensvollen Daten könnte ein großer Schritt in Richtung einer weiteren Demokratisierung der Gesellschaft werden. Viele Falschnachrichten könnten leicht von jedermann durch entsprechende Fakten oder statistischen Auswertungen paralysiert werden.
Vielleicht kann man die Schaffung einer solchen Programmiersprache mit der Schaffung des ersten Alphabets durch die Phönizier oder der Schaffung des ersten Alphabets mit Vokalen durch die Griechen vergleichen. Hätten diese Völker solche Leistungen vollbringen können ohne diese Voraussetzungen. Ich vermute ohne dieses Alphabet hätte es keine griechische Wissenschaft und Kultur gegeben; vielleicht auch keine griechische Demokratie.
Entwurfskriterien für eine solche Sprache:
- Eine mathematische Fundierung ist erforderlich.
- Methodisch-didaktische und pragmatische Fragen stehen zunächst vor Effizienzproblemen.
- Kurze, lesbare Programme; die wichtigsten Schlüsselworte sollten kurz sein
- Einfache, unstrukturierte Programme; Schleifen und allgemeine Rekursionen führen häufig zu schwer lesbaren und schwer änderbaren Programmen;
- Universelle Anwendbarkeit; sie muss nicht nur für Relationen (flache einfache Tabellen) sondern auch für strukturierte Tabellen und Dokumente nutzbar sein; sie muss nicht nur für Anfragen an die wichtigsten Systeme sondern auch für vielfältige Berechnungen geeignet sein
- Um im Schulunterricht einsetzbar zu sein, muss sie die verschiedenen mathematische Teilgebiete unterstützen, sowie Nutzen für die anderen Fächer bieten
- Sie sollte so mächtig sein, dass sie andere Systeme und Sprachen wie Tabellenkalkulation und SQL ersetzen kann.
- Aus Endnutzersicht darf es nur ein einheitliches System mit einheitlicher Syntax (Schreibweise) für die Verarbeitung von Massendaten geben, genau wie die Operationen der Einzeldatenverarbeitung (+ – * : sin) standardisiert sind.
Einführung in o++o:
A. Merkel „Jeder Schüler soll neben lesen, rechnen und schreiben auch programmieren können.“
o++o (ausführlich ottoPS) ist eine tabellenorientierte Programmiersprache mit funktionalen Möglichkeiten, die auf Schleifen verzichtet. Dennoch ist o++o sehr ausdrucksstark und man kann mit ihr nicht nur kompakte Anfragen sondern auch vielfältige Berechnungen für strukturierte Tabellen und strukturierte Dokumente bewerkstelligen.
o++o benutzt viele mathematische Konzepte, daher sehen wir die Hauptvorteile der Vermittlung im Mathematikunterricht, genau wie die wesentlichen Fähigkeiten für die Nutzung des Taschenrechners in Mathematik vermittelt werden. o++o verwendet insbesondere folgende Konzepte: Kollektion (Menge, Multimenge, Liste); Gleichheit und Inklusionsbeziehungen dieser; Tupel; leistungsfähige Operationen zum Selektieren; Berechnen; Restrukturieren; Sortieren und Aggregieren (Summe; Durchschnitt; …),… .
Tabellenkalkulationsprogramme wie EXCEL und die Datenbankstandardabfragesprache SQL kennen keine strukturierten Schemen und Tabellen. Erste Tests mit Vorschulkindern lassen vermuten, dass man mit strukturierten Tabellen leichter rechnen kann als mit Dezimalzahlen. Wir wollen einige o++o-Beispielprogramme anfügen:
1. Berechne den Wert eines einfachen Terms.
2*3+4
* und + haben jeweils 2 Inputwerte. Zunächst wird 2*3 (6) berechnet. Die 6 ist erster Inputwert von +, so dass sich insgesamt 24 ergibt. Hier wird also einfach von links nach rechts gerechnet.
2. Schreibe den Term cos³(sin²(3.14159)) in o++o.
pi sin hoch 2 cos hoch 3
Unserer Meinung nach ist der Ausgangsterm für Otto Normalverbraucher schwer zu lesen. Man beginnt mit pi geht nach links bis zum sin dann nach rechts zum hoch 2 jetzt bewegt man sich wieder nach links zum cos und abschließend nach rechts zum hoch 3. Diese Schreibweise wurde sicher eingeführt um Klammern zu sparen. Eigentlich müsste der Ausgangsterm um unmissverständlich zu sein, folgendes Aussehen haben:
(cos((sin(3.14159))²))³
Das ist sicher noch schwerer zu lesen und man bewegt sich noch mehr von links nach rechts und umgekehrt.
3. Schreibe den Term sin²(x)+cos³(y) in o++o.
X sin hoch 2 + (Y cos hoch 3)
oder
X sin hoch 2
+ Y cos hoch 3
Man könnte alle Terme in o++o ohne Klammern schreiben, allerdings müssten dann bestimmte Terme mehrzeilig geschrieben werden.
4. Wie berechnet man den Term 2+3:4*5 ?
2+(3:(4*5))=2 3/20
2+((3:4)*5)=5 ¾
o++o: ((2+3):4)*5=6 1/4
Man erkennt, dass man mit der Schulweisheit Punktrechnung geht vor Strichrechnung noch nicht auskommt. Man benötigt die Regel „von links nach rechts“ zusätzlich.
5. Berechne den Durchschnitt mehrerer Noten.
1 2 3 1 2 ++:
Vom methodischen Standpunkt kann man dieses Programm noch verbessern, indem man die Klammern für Listen hinzufügt: [1 2 3 1 2] ++:
Man erkennt jetzt, dass die Durchschnittsoperation ++: einen Inputwert, nämlich eine Liste besitzt und dass ++: diesem einen Inputwert nachgestellt wird. Da die Nutzer in der Regel nicht viel tippen wollen, gehen wir davon aus, dass die erste Notation in Praxis häufiger benutzt werden wird.
6. Berechne die Durchschnitte einer strukturierten Tabelle noten.tab für jedes Fach.
noten.tab
DUR:=NOTEl ++:
noten.tab könnte so aussehen:
FACH,NOTEl l
Ma 1 2 1 3 1 2
Phy 4 3 2 2 1
…
Hierbei kürzt l Liste ab. D.h., noten.tab ist eine einfache strukturierte Tabelle (Liste), die zu jedem Fach eine Liste von Noten enthält. Um Platz zu sparen, wählen wir auch hier die methodisch nicht optimale Darstellung. Wie FACH ist auch NOTE ein Spaltenname, so dass noten.tab eigentlich so dargestellt werden müsste:
FACH,NOTEl l
Ma 1 2 1 3 1 2
Phy 4 3 2 2 1
Das Ergebnis der Anfrage wieder im „tab-Format“:
FACH, DUR, NOTEl l
Ma 1.66666666667 1 2 1 3 1 2
Phy 2.4 4 3 2 2 1
7. Bilde die Summe der Zahlen von 1 bis 100 (Aufgabe von Gauß Klasse 5).
1 .. 100 ++
Wie die Addition und die Multiplikation besitzt .. zwei Inputwerte (1 und 100). Als Zwischenergebnis entsteht die Liste
ZAHLl
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23
24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43
44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63
64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83
84 85 86 87 88 89 90 91 92 93 94 95 96 97 98 99 100
,deren Zahlen dann aufsummiert werden, so dass sich 5050 ergibt.
8. Berechne näherungsweise das Maximum der Sinus-Funktion im Intervall [1 2].
1 … 2!0.001 sin max
… benötigt 3 Inputwerte: 1. den Anfangswert 1, den Endwert 2 und die Schrittweite 0.001. Es entstehen hierbei die Zahlen 1 1.001 1.002 1.003 …1.999 2.
Auf jede der Zahlen wird die Sinusfunktion angewandt, sodass wieder 1001 Zahlen entstehen. Auf diese Liste wird dann die Funktion max (Maximum) angewandt. Obwohl es sich hierbei um ein Näherungsverfahren handelt, kommt der exakte Wert 1 heraus, wenn die Schrittweite weiter verfeinert wird. sin und max haben jeweils einen Inputwert (hier eine Liste) aber der Outputwert von sin ist wieder eine Liste und max erzeugt lediglich eine Zahl, da es sich hier um eine Aggregationsfunktion handelt. Der zweite und der dritte Inputwert einer dreistelligen Operation (oben …) wird jeweils durch ein „!“ getrennt. Das ist in o++o nötig, da das Komma für die Paarbildung bereits vergeben ist und das Leerzeichen bereits Listenelemente trennt.
9. Berechne näherungsweise das Minimum des Polynoms X³ + 4 X² -3 X+2 im Intervall [0 2] mit zugehörigem X-Wert.
[X! 0 … 2!0.001]
Y:= X polynom [1 4 -3 2]
MINI:= Yl min
avec Y = MINI
avec ist französisch und bezeichnet eine Selektion. Ein konkretes Polynom von einer Variablen X hat stets nur einen Inputwert, der für X eingesetzt wird. polynom in Zeile 2 ist dagegen allgemeiner und hat 2 Inputwerte:
- Den Inputwert für X, der hier alle Zahlen, die in der ersten Zeile generiert wurden, annimmt.
- Eine Liste von Zahlen, die den Koeffizienten des konkreten Polynoms entspricht.
Durch die ersten Zeile entsteht eine Liste von Zahlen, die alle den Namen X bekommen haben. Das erkennt man am besten in der xml bzw. ment-Repräsentation:
<X>0.</X>
<X>0.001</X>
<X>0.002</X>
Gesamtergebnis:
MINI, (X, Y l)
1.481482037 0.333 1.481482037
10. Berechne eine Nullstelle der Cosinus Funktion im Intervall [1 2] näherungsweise.
[X! 1 … 2!0.0001]
avec X cos < 0
avec X pos = 1
Hier verbleiben nach der ersten Selektion nur die X-Werte mit Funktionswert kleiner 0. Von diesen wird im zweiten Schritt der erste Wert ausgewählt. Da wir wissen, dass cos nur eine Nullstelle im betrachteten Intervall besitzt, wird diese durch das Ergebnis angenähert. pos kürzt Position ab, so dass das erste Paar der verbliebenen Paare selektiert wird.
11. Berechne das Gesamtwachstum, wenn 5 Jahreswachstumszahlen gegeben sind. Runde das Ergebnis auf eine Stelle nach dem Komma.
[W! 0 1.5 2.1 1.3 0.4 1.2]
ACCU:= first 100. next ACCU pred *(W:100+1) at W
rnd 1
Die Ergebnistabelle:
[W! 0 1.5 2.1 1.3 0.4 1.2]
ACCU:= first 100. next ACCU pred *(W:100+1) at W
rnd 1
Die Ergebnistabelle:
W, ACCU l
0. 100.
1.5 101.5
2.1 103.6
1.3 105.
0.4 105.4
1.2 106.7
Der erste ACCU-Wert ergibt sich durch den Ausdruck hinter first (100.). Für den zweiten Wert wird für ACCU pred der Wert 100. eingesetzt und der Term nach next bewertet. Es ergibt sich 101.5. Diese Zahl wird wieder in ACCU pred eingesetzt und der next-Term erneut berechnet (rund 103.6),… bis der letzte W-Wert erreicht ist. pred ist der predecessor (Vorgänger).
12. Berechne die Fläche unter der Sinuskurve im Intervall [0, pi] näherungsweise.
0 … pi!0.0001 sin * 0.0001 ++
Hierbei werden nacheinander alle Zahlen zwischen 0 und pi generiert, dann von jeder Zahl der Sinus berechnet und anschließend jede Zahl mit 0.0001 multipliziert. Es entstehen 31415 Rechteckflächen, die abschließend addiert werden.
13. Berechne den DurchschnittsBMI pro Alter und den BMI pro Person und Alter für alle Personen über 20.
<TAB!
NAME, LAENGE, (ALTER, GEWICHT l) l
Klaus 1.68 18 61 30 65 56 80
Rolf 1.78 40 72
Kathi 1.70 18 55 40 70
Walleri 1.00 3 16
Viktoria 1.61 13 51
Bert 1.72 18 66 30 70
!TAB>
avec NAME! 20<ALTER
BMI:= GEWICHT : LAENGE : LAENGE
gib ALTER,BMIAVG,(NAME,BMI m) m BMIAVG:= BMI ! ++:
rnd 2 #rundet alle Zahlen der Tabelle auf 2 Stellen nach dem Punkt
Die TAB-Klammern deuten an, dass die eingeschlossenen Daten der TAB-Darstellung entsprechen.
Die obige Bedingung selektiert Personen-Sätze, d.h. NAME,LAENGE,(ALTER,GEWICHT l) Tupel (strukturierte Tupel bzw. Strupel). Da eine Personen mehrere ALTER-Angaben besitzt, muss quantifiziert werden. NAME! 20 <ALTER selektiert demnach alle Personen, die einen entsprechenden Alterseintrag besitzen. D.h., der Existenzquantor wird nicht geschrieben, gehört aber zu jeder Bedingung. In diesem kleinen Beispiel könnte man die Selektion natürlich auch per Hand realisieren.
Resultat:
ALTER, BMIAVG, (NAME, BMI m) m
18 20.98 Bert 22.31
Kathi 19.03
Klaus 21.61
30 23.35 Bert 23.66
Klaus 23.03
40 23.47 Kathi 24.22
Rolf 22.72
56 28.34 Klaus 28.34
Das Endergebnis kann beispielsweise durch einfaches Klicken als Säulendiagramm dargestellt werden. Das Beispiel zeigt, dass man eine Hierarchie einfach durch Angabe des gewünschten Schemas umkehren kann. Im Ergebnis ist der Name dem Alter untergeordnet.
Es wird insbesondere deutlich, dass die Aufgaben ohne Kenntnisse der Differential- und Integral-rechnung gelöst werden können. Mit o++o kann der Mathematikunterricht in vielfältiger Weise unterstützt werden. Das reicht von Klasse 7 oder tiefer bis zur Klassenstufe 12. Es betrifft: Rechnen mit natürlichen Zahlen, Dezimalzahlen, näherungsweise Berechnung von Nullstellen beliebiger Funktionen, Ableitung, Flächen unter Kurven, Extremwerte (kann wahrscheinlich bereits in der Sekundarschule gelehrt werden), Wahrscheinlichkeitsrechnung, … . Mit o++o können Dinge in einfacher Weise berechnet werden, die sonst nur theoretisch abgehandelt werden. Dadurch kann das Verständnis der Konzepte wesentlich verbessert, erweitert und vertieft werden. Weitere Informationen zu o++o finden Sie unter ottops.de (Z.B. „o++o auf 8 Seiten“ ist eine kurze Einführung).
Wir glauben, dass o++o besondere Vorteile für den Mathematik- und Informatikunterricht bietet aber auch in den anderen Fächern sinnvoll genutzt werden kann.
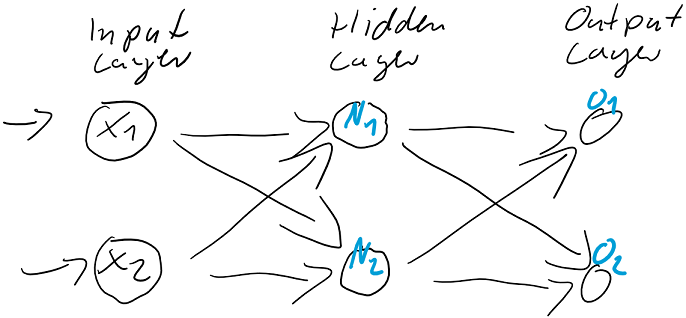
 berechnen…
berechnen…


 werden dann als Berechnungsgrundlage für die Ausgaben der Ausgabeschicht
werden dann als Berechnungsgrundlage für die Ausgaben der Ausgabeschicht  verwendet. Auch die Ausgabe-Neuronen berechnen ihre jeweilige Nettoeingabe
verwendet. Auch die Ausgabe-Neuronen berechnen ihre jeweilige Nettoeingabe 
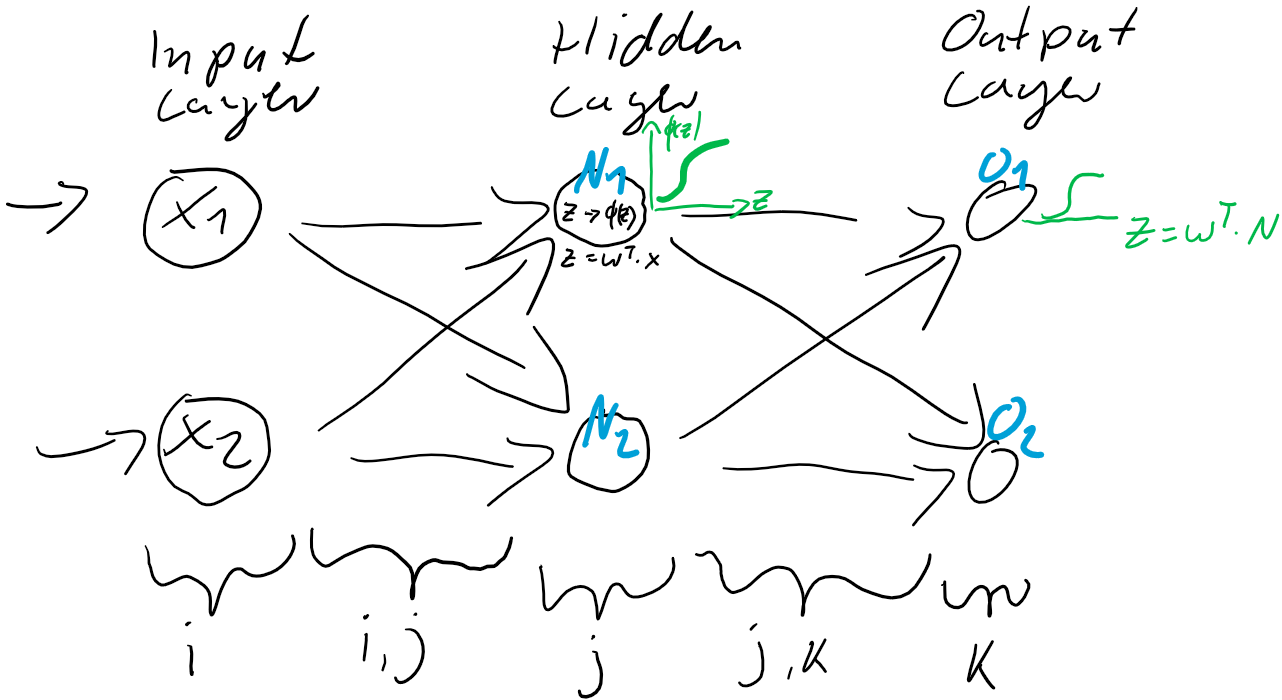
 ) und der Prädiktion (Ausgabe
) und der Prädiktion (Ausgabe 
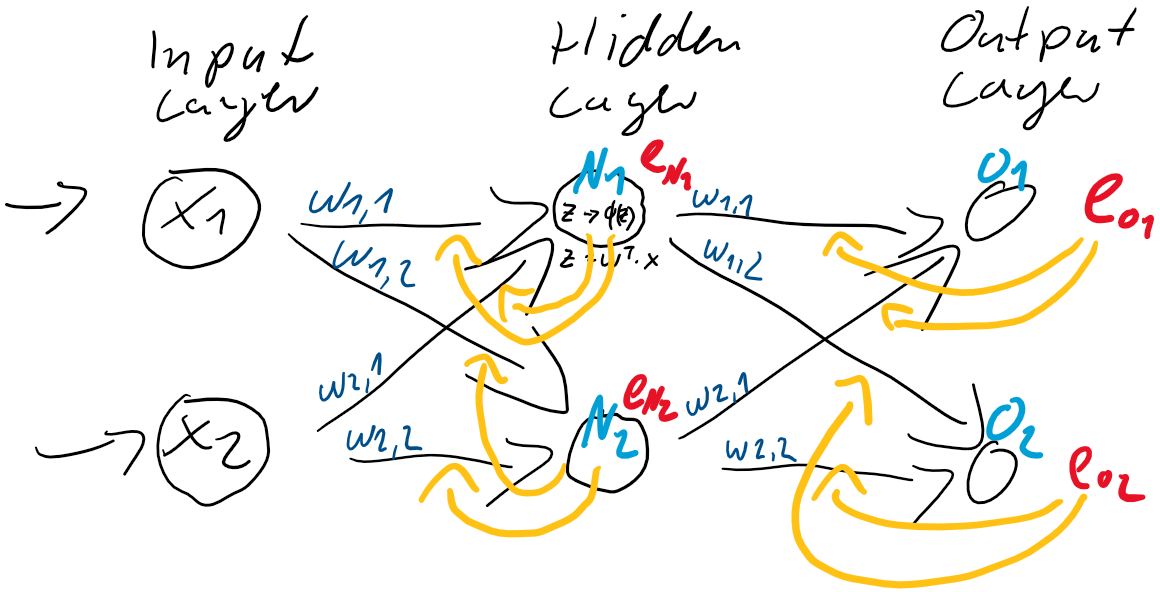
 ist also einfach der Unterschied zwischen dem Ziel-Wert und der Prädiktion. Jedes Training ist eine Wiederholung von Prädiktion (Forward) und Gewichtsanpassung (Back). Im ersten Schritt werden üblicherweise die Gewichtungen zufällig gesetzt, jede Gewichtung unterschiedlich nach Zufallszahl. So ist die Wahrscheinlichkeit, gleich zu Beginn die “richtigen” Gewichtungen gefunden zu haben auch bei kleinen neuronalen Netzen verschwindend gering. Der Fehler wird also groß sein und kann über den Gradientenabstieg durch Gewichtsanpassung verkleinert werden.
ist also einfach der Unterschied zwischen dem Ziel-Wert und der Prädiktion. Jedes Training ist eine Wiederholung von Prädiktion (Forward) und Gewichtsanpassung (Back). Im ersten Schritt werden üblicherweise die Gewichtungen zufällig gesetzt, jede Gewichtung unterschiedlich nach Zufallszahl. So ist die Wahrscheinlichkeit, gleich zu Beginn die “richtigen” Gewichtungen gefunden zu haben auch bei kleinen neuronalen Netzen verschwindend gering. Der Fehler wird also groß sein und kann über den Gradientenabstieg durch Gewichtsanpassung verkleinert werden. und
und  und passen danach die Gewichte
und passen danach die Gewichte  (
( &
&  und
und  &
&  ) der Schicht zwischen dem Hidden-Layer
) der Schicht zwischen dem Hidden-Layer 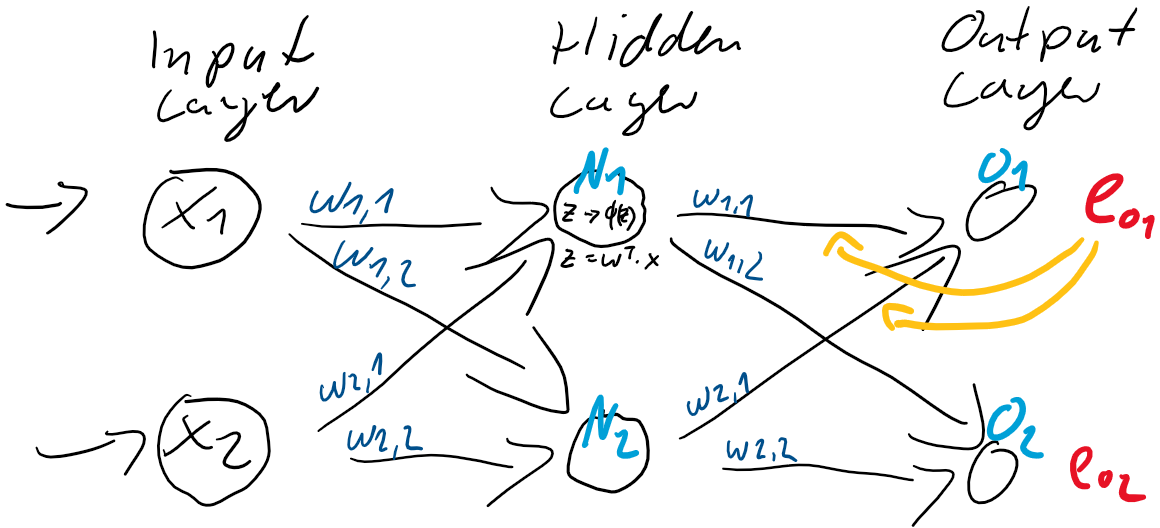
 und dem Hidden-Layer
und dem Hidden-Layer  und
und  . Dieser Anteil am Fehler der jeweiligen Neuronen ergibt sich direkt aus den Gewichtungen
. Dieser Anteil am Fehler der jeweiligen Neuronen ergibt sich direkt aus den Gewichtungen 

![\[ e_{N} = \left(\begin{array}{rr} \frac{w_{1,1}}{w_{1,1} + w_{1,2}} & \frac{w_{1,2}}{w_{1,1} + w_{1,2}} \\ \frac{w_{2,1}}{w_{2,1} + w_{2,2}} & \frac{w_{2,2}}{w_{2,1} + w_{2,2}} \end{array}\right) \cdot \left(\begin{array}{c} e_{1} \\ e_{2} \end{array}\right) \qquad \]](https://data-science-blog.com/de/wp-content/ql-cache/quicklatex.com-6fadfe69caec8e8c351788824875138a_l3.png "Rendered by QuickLaTeX.com")
![\[ e_{N} = \left(\begin{array}{rr} w_{1,1} & w_{1,2} \\ w_{2,1} & w_{2,2} \end{array}\right) \cdot \left(\begin{array}{c} e_{1} \\ e_{2} \end{array}\right) \qquad \]](https://data-science-blog.com/de/wp-content/ql-cache/quicklatex.com-7dee7d02ac1aba606a45895afe90a837_l3.png "Rendered by QuickLaTeX.com")

 zwischen der Eingabe-Schicht
zwischen der Eingabe-Schicht  und der verborgenden Schicht
und der verborgenden Schicht  .
.

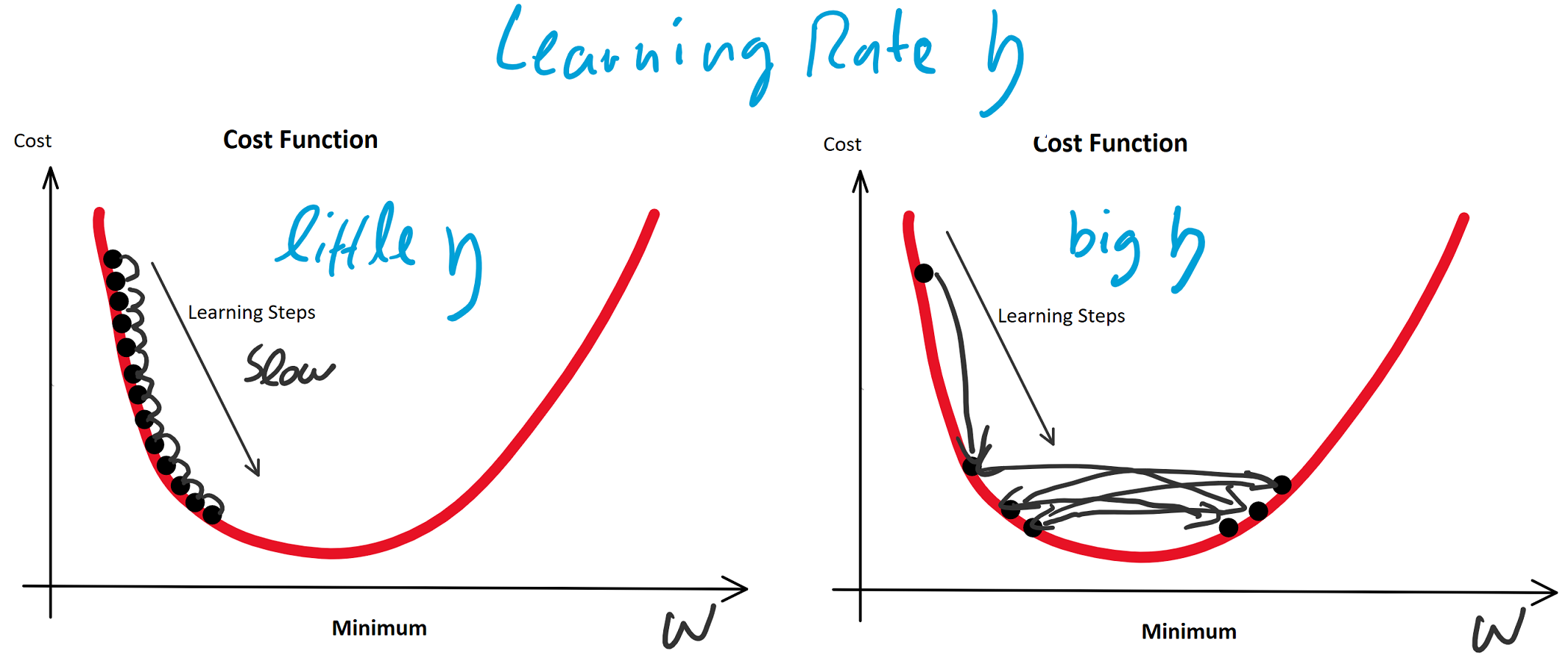
 (Random Initialization). Wir berechnen die Ausgabe (Forwardpropogation) und vergleichen sie über eine Verlustfunktion (z. B. über die Funktion Mean Squared Error) mit dem tatsächlich korrekten Wert. Auf Grund der zufälligen Initialisierung haben wir eine nahe zu garantierte Falschheit der Ergebnisse und somit einen Verlust. Für die Verlustfunktion berechnen wir den Gradienten für gegebene Eingabewerte. Voraussetzung dafür ist, dass die Funktion ableitbar ist. Wir bewegen uns entgegen des Gradienten in Richtung Minimum der Verlustfunktion. Ist dieses Minimum (fast) gefunden, spricht man auch davon, dass der Lernalgorithmus konvergiert.
(Random Initialization). Wir berechnen die Ausgabe (Forwardpropogation) und vergleichen sie über eine Verlustfunktion (z. B. über die Funktion Mean Squared Error) mit dem tatsächlich korrekten Wert. Auf Grund der zufälligen Initialisierung haben wir eine nahe zu garantierte Falschheit der Ergebnisse und somit einen Verlust. Für die Verlustfunktion berechnen wir den Gradienten für gegebene Eingabewerte. Voraussetzung dafür ist, dass die Funktion ableitbar ist. Wir bewegen uns entgegen des Gradienten in Richtung Minimum der Verlustfunktion. Ist dieses Minimum (fast) gefunden, spricht man auch davon, dass der Lernalgorithmus konvergiert.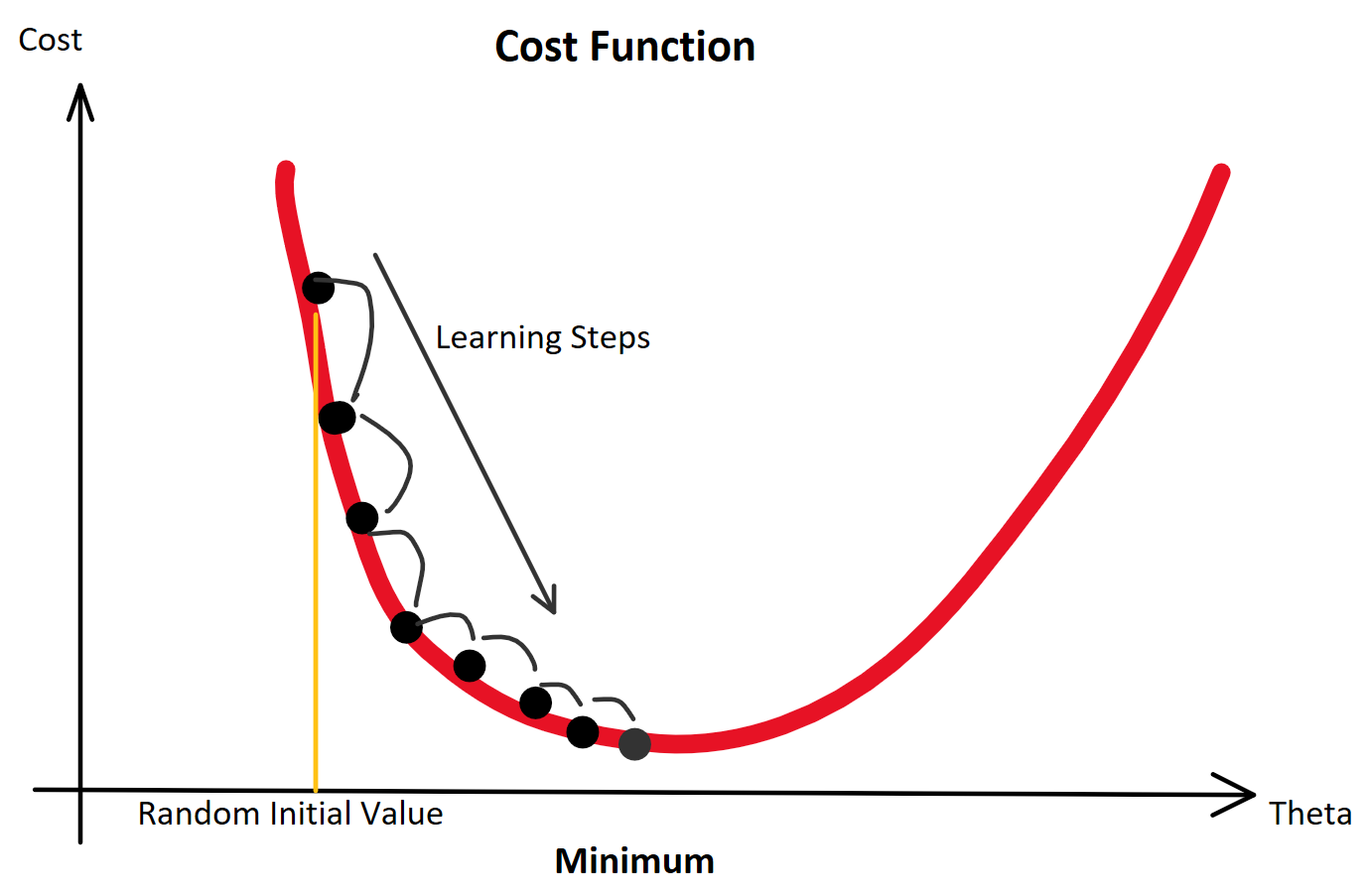
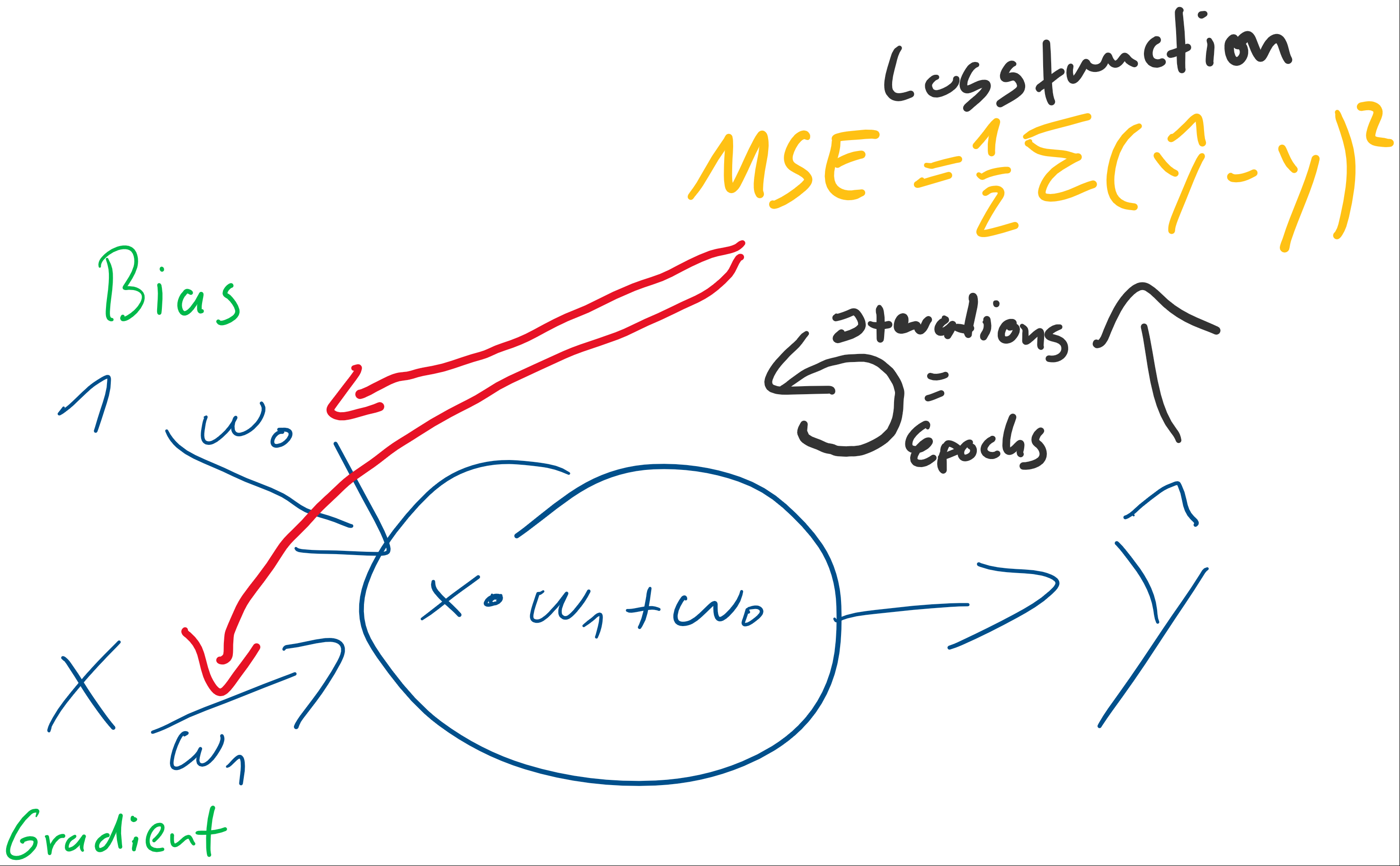
 ) der stets mit einer Eingangskonstante multipliziert und somit als Wert erhalten bleibt. Der Bias ist das Alpha
) der stets mit einer Eingangskonstante multipliziert und somit als Wert erhalten bleibt. Der Bias ist das Alpha  in einer Schulmathe-tauglichen Formel wie
in einer Schulmathe-tauglichen Formel wie  .
. ist die Steigung, der Gradient, der Funktion.
ist die Steigung, der Gradient, der Funktion. und den darauffolgenden Abgleich mit dem tatsächlichen
und den darauffolgenden Abgleich mit dem tatsächlichen  herauszufinden. Anfangs behaupten wir beispielsweise einfach, sowohl
herauszufinden. Anfangs behaupten wir beispielsweise einfach, sowohl  . Folglich wird
. Folglich wird 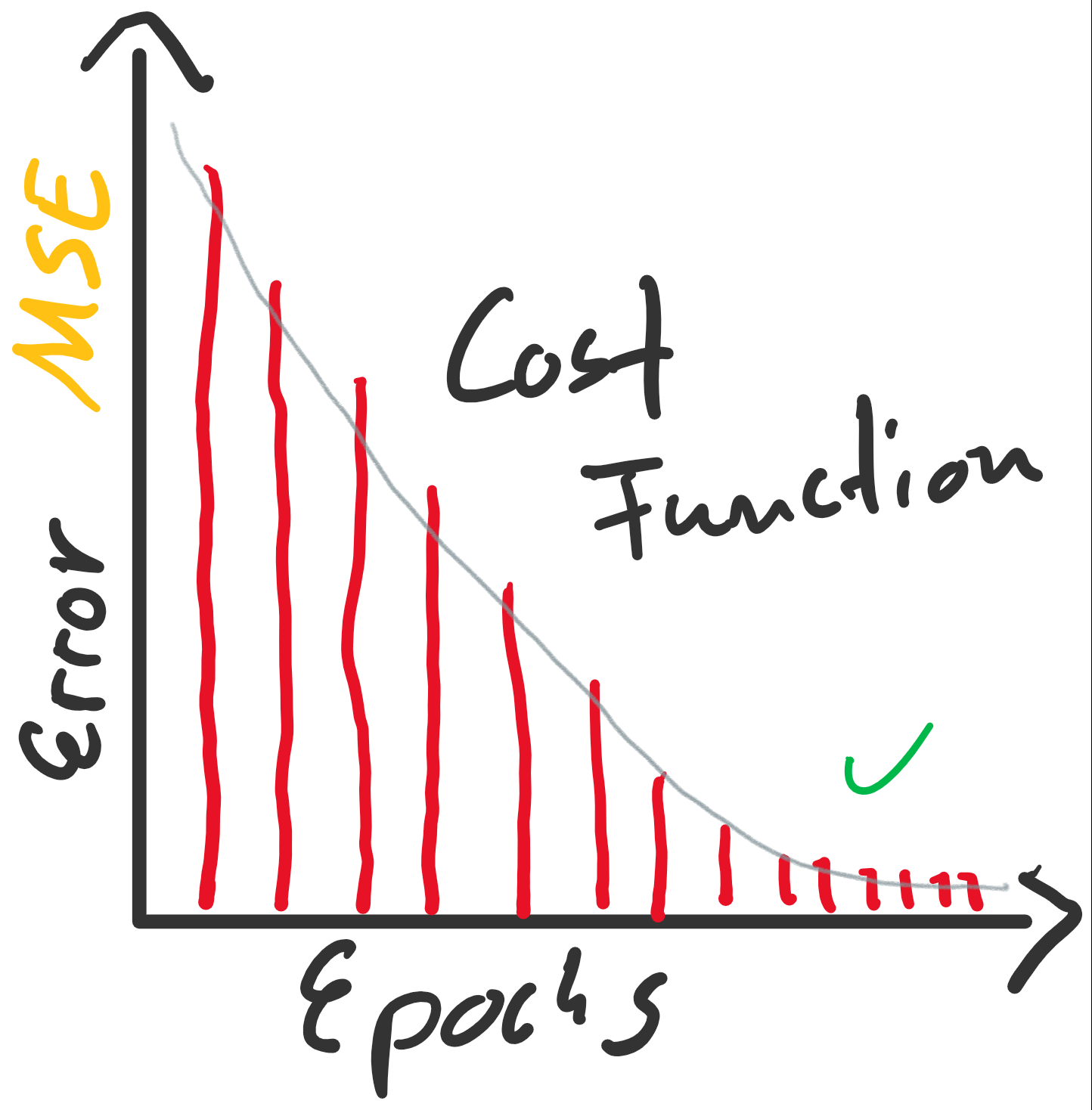



 bzw. partiell nach jedem vorhandenen
bzw. partiell nach jedem vorhandenen  :
:


 am Ende her haben? Das ergibt sie aus der Kettenregel: Die äußere Funktion wurde abgeleitet, so wurde aus
am Ende her haben? Das ergibt sie aus der Kettenregel: Die äußere Funktion wurde abgeleitet, so wurde aus  dann
dann  . Jedoch muss im Sinne eben dieser Kettenregel auch die innere Funktion abgeleitet werden. Da wir nach
. Jedoch muss im Sinne eben dieser Kettenregel auch die innere Funktion abgeleitet werden. Da wir nach  erhalten.
erhalten.
 ist der Gradient der Funktion!)
ist der Gradient der Funktion!)
 (eta) bezeichnet:
(eta) bezeichnet:
 enthalten sind. Der Fehler ist dann aber sehr groß (sollte maximal sein, im Vergleich zu zukünftigen Epochen). Die Gewichte werden also angepasst, die Gerade somit besser in die Punktwolke platziert. Mit jeder Epoche wird die Gerade erneut in die Punktwolke gelegt, der Gesamtfehler (über alle
enthalten sind. Der Fehler ist dann aber sehr groß (sollte maximal sein, im Vergleich zu zukünftigen Epochen). Die Gewichte werden also angepasst, die Gerade somit besser in die Punktwolke platziert. Mit jeder Epoche wird die Gerade erneut in die Punktwolke gelegt, der Gesamtfehler (über alle 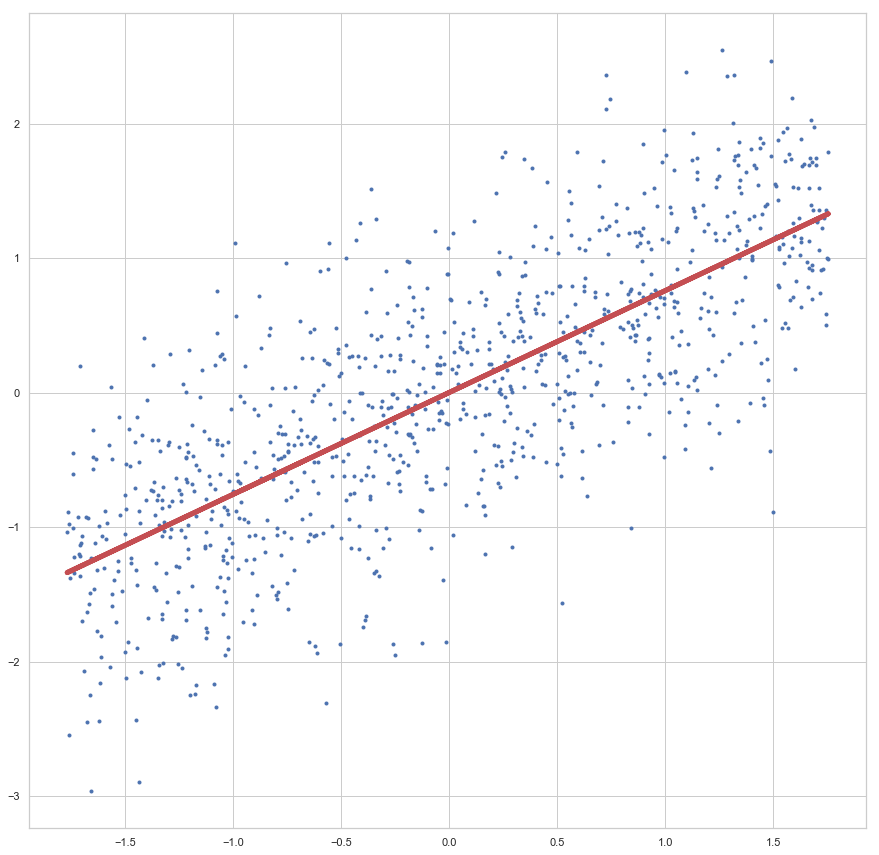
 konvergiert:
konvergiert: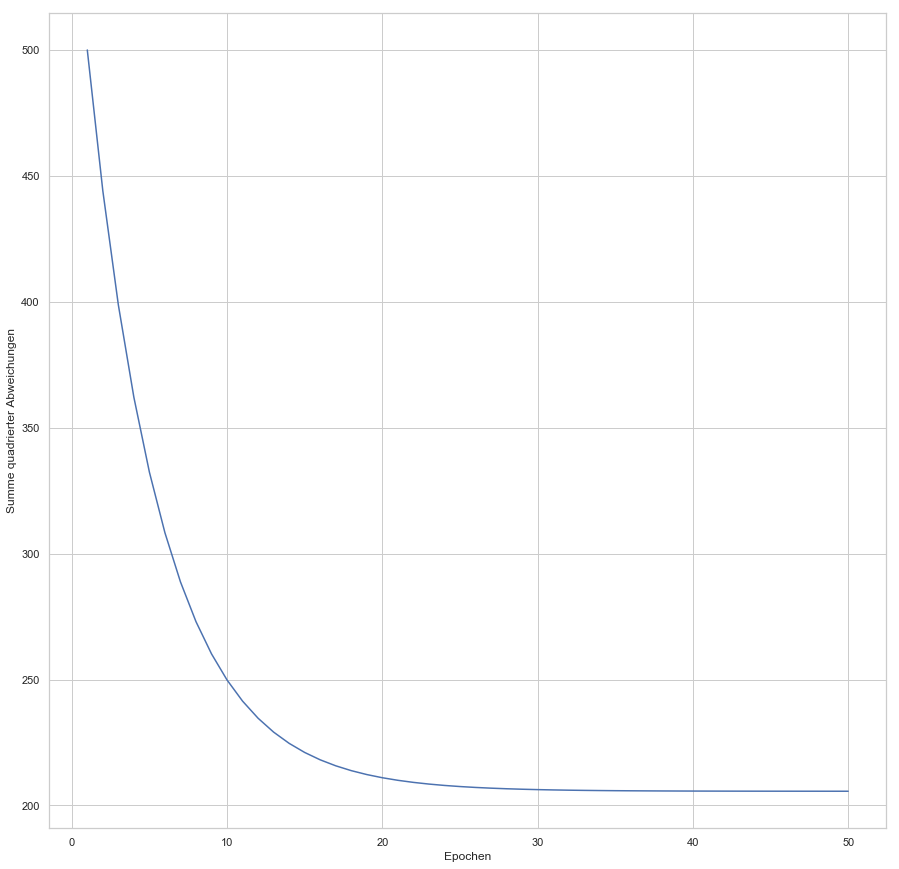


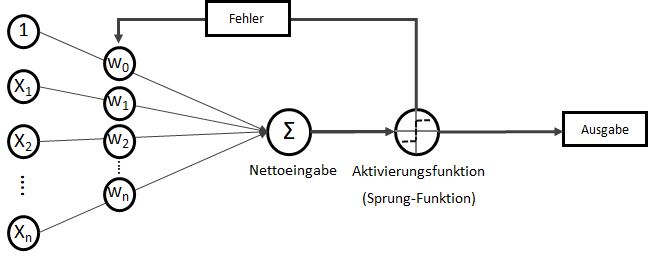
 . Wobei für
. Wobei für  als Bias-Input stets gilt:
als Bias-Input stets gilt:  . Der Bias-Input ist nur ein Platzhalter für das wichtige Bias-Gewicht.
. Der Bias-Input ist nur ein Platzhalter für das wichtige Bias-Gewicht.![\[ x = \begin{bmatrix} x_0\\ x_1\\ x_2\\ x_3\\ \vdots\\ x_n \end{bmatrix} \]](https://data-science-blog.com/de/wp-content/ql-cache/quicklatex.com-3a7aa03dea498a7231e3e497e3e5673d_l3.png "Rendered by QuickLaTeX.com")

![\[ w = \begin{bmatrix} w_0\\ w_1\\ w_2\\ w_3\\ \vdots\\ w_n \end{bmatrix} \]](https://data-science-blog.com/de/wp-content/ql-cache/quicklatex.com-c70ad1733451ddc4512b23f2c739f80e_l3.png "Rendered by QuickLaTeX.com")
 mit
mit  .
.![\[ z = w_0 \cdot x_0 + w_1 \cdot x_1 + \dots + w_n \cdot x_n \]](https://data-science-blog.com/de/wp-content/ql-cache/quicklatex.com-3638f2ae3b558db6c95e18057b1b9898_l3.png "Rendered by QuickLaTeX.com")
 mit der Eingabe
mit der Eingabe 
 .
.![\[ z = w^T \cdot x \]](https://data-science-blog.com/de/wp-content/ql-cache/quicklatex.com-fc9f71e279d7fd0095c17065743df909_l3.png "Rendered by QuickLaTeX.com")
 steht dabei für transponieren. Transponieren bedeutet, dass Spalten zu Zeilen werden – oder umgekehrt.
steht dabei für transponieren. Transponieren bedeutet, dass Spalten zu Zeilen werden – oder umgekehrt.![\[ x = \begin{bmatrix} 5\\ 12\\ 30\\ 2 \end{bmatrix} \]](https://data-science-blog.com/de/wp-content/ql-cache/quicklatex.com-97145f06e658d2f10086bd8c80293749_l3.png "Rendered by QuickLaTeX.com")
![\[ w = \begin{bmatrix} 1\\ 2\\ 5\\ 12 \end{bmatrix} \]](https://data-science-blog.com/de/wp-content/ql-cache/quicklatex.com-6cd08615346cc16d7e8c23bac32c19b9_l3.png "Rendered by QuickLaTeX.com")
![\[ z = w^T \cdot x = \big[1\text{ }2\text{ }5\text{ }12\big] \cdot \begin{bmatrix} 5\\ 12\\ 30\\ 2 \end{bmatrix} = 1 \cdot 5 + 2 \cdot 12 + 5 \cdot 30 + 12 \cdot 2 = 203 \]](https://data-science-blog.com/de/wp-content/ql-cache/quicklatex.com-70a3e9e9243199e97c549d1d9b0f44c0_l3.png "Rendered by QuickLaTeX.com")
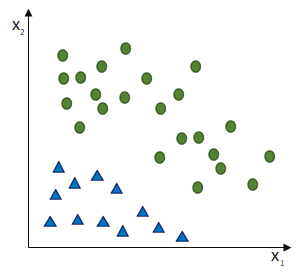
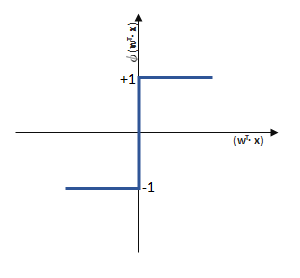
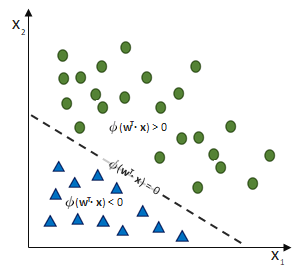


 entgegen des Fehlers (bzw. hin zur jeweils anderen möglichen Antwort) geschieht:
entgegen des Fehlers (bzw. hin zur jeweils anderen möglichen Antwort) geschieht:
 ausgehen.
ausgehen.







 ist und das SLP irrtümlicherweise die Klasse
ist und das SLP irrtümlicherweise die Klasse  ausgewiesen hat, obwohl die korrekte Klasse
ausgewiesen hat, obwohl die korrekte Klasse  wäre. (Und die Schrittweite lassen wir bei
wäre. (Und die Schrittweite lassen wir bei  )
)
 verringert sich entsprechend
verringert sich entsprechend  und somit wird die Wahrscheinlichkeit größer, dass wenn bei der nächsten Iteration (
und somit wird die Wahrscheinlichkeit größer, dass wenn bei der nächsten Iteration ( ) wieder die Klasse +1 korrekt sei, den Schwellwert
) wieder die Klasse +1 korrekt sei, den Schwellwert  zu unterschreiten und auf eben diese korrekte Klasse zu stoßen.
zu unterschreiten und auf eben diese korrekte Klasse zu stoßen. . So würde beispielsweise ein neues
. So würde beispielsweise ein neues  (bei Iteration
(bei Iteration  ) zu einer irrtümlichen Klassifikation
) zu einer irrtümlichen Klassifikation  (
( ) führen, würde die Entscheidungsgrenze zur korrekten Prädiktion der Klasse beim nächsten Durchlauf (
) führen, würde die Entscheidungsgrenze zur korrekten Prädiktion der Klasse beim nächsten Durchlauf ( ) an
) an 
 und
und 
 funktioniert.
funktioniert. springt, wenn z > 0 ist, ansonsten aber
springt, wenn z > 0 ist, ansonsten aber  bleibt.
bleibt.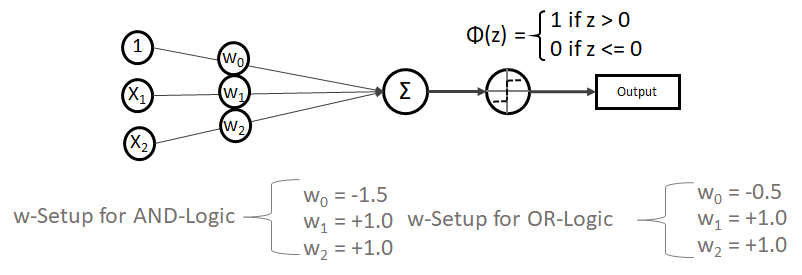
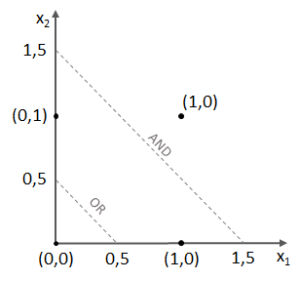
 ,
,
 ,
,
 ,
,
 ,
, ,
, ,
,

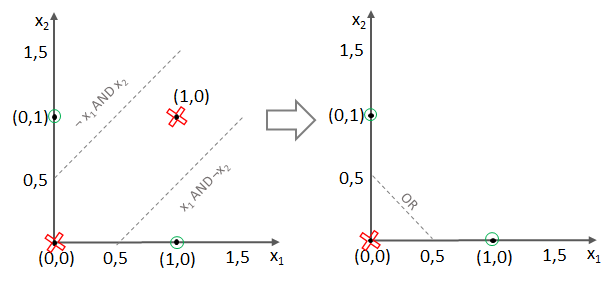
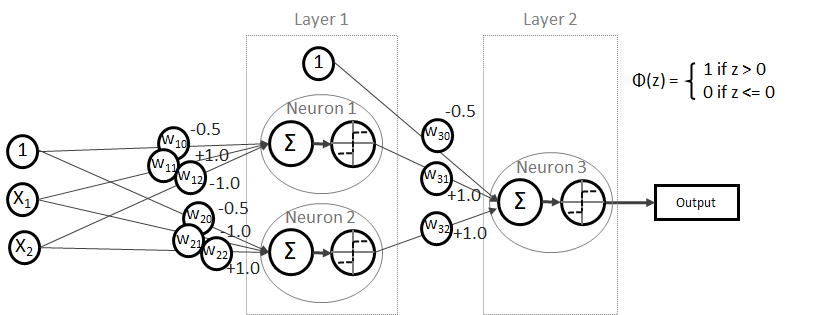
 und somit
und somit 
 und somit
und somit 
 und somit
und somit 
 und somit
und somit 
 und somit
und somit 
 und somit
und somit 
 und somit
und somit 
 und somit
und somit 
 und somit
und somit  und somit
und somit  und somit
und somit 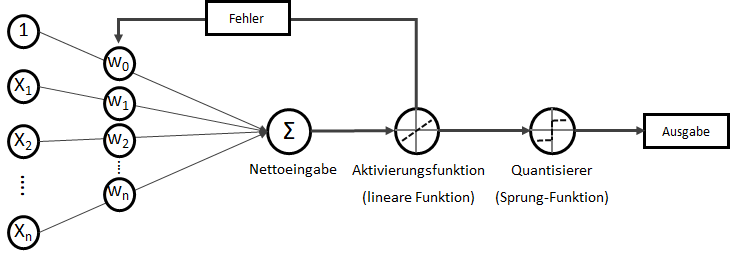







 kann allgemein mit folgender Formel bestimmt werden
kann allgemein mit folgender Formel bestimmt werden  .
. aus dem Datenraum
aus dem Datenraum  , die ein Trainer dem Lernverfahren vorgibt, um Zielkonzept c zu erlernen, eine Hypothese aus dem Hypothesenraum
, die ein Trainer dem Lernverfahren vorgibt, um Zielkonzept c zu erlernen, eine Hypothese aus dem Hypothesenraum  des Lernverfahrens zu ermitteln, welche (möglichst) alle positiven Beispiel
des Lernverfahrens zu ermitteln, welche (möglichst) alle positiven Beispiel  umfasst und (möglichst) alle negativen Beispiele
umfasst und (möglichst) alle negativen Beispiele  ausschließt.
ausschließt.
 .
. und im Fall von Dummy-Encoding
und im Fall von Dummy-Encoding  neue Bool’sche Eigenschaften.
neue Bool’sche Eigenschaften. und für allgemeine endliche Eigenschaften
und für allgemeine endliche Eigenschaften . Diese Repräsentation ist sehr eingeschränkt und erlaubt es nur einzelne und keine kombinierten Konzepte zu erlernen. Sie ist daher eigentlich nur von theoretischem Interesse und wird – soweit bekannt – in keinem praktisch eingesetzten Lernverfahren genutzt.
. Diese Repräsentation ist sehr eingeschränkt und erlaubt es nur einzelne und keine kombinierten Konzepte zu erlernen. Sie ist daher eigentlich nur von theoretischem Interesse und wird – soweit bekannt – in keinem praktisch eingesetzten Lernverfahren genutzt. . Mit dieser Sprache können alle Eigenschaften zwar separat auf beliebige Teilmengen generalisiert werden, Korrelationen zwischen Eigenschaften werden jedoch nicht berücksichtigt.
. Mit dieser Sprache können alle Eigenschaften zwar separat auf beliebige Teilmengen generalisiert werden, Korrelationen zwischen Eigenschaften werden jedoch nicht berücksichtigt. . Auf beliebige endliche Eigenschaften übertragen, kann diese Aussage zu
. Auf beliebige endliche Eigenschaften übertragen, kann diese Aussage zu  verallgemeinert werden.
verallgemeinert werden.

 die ein „konsistenter Lernalgorithmus“
die ein „konsistenter Lernalgorithmus“ und einer Unsicherheit
und einer Unsicherheit  (bzw. einer Wahrscheinlichkeit von
(bzw. einer Wahrscheinlichkeit von  ) zu erlernen, abgeschätzt werden mit
) zu erlernen, abgeschätzt werden mit![\[m \geq \frac{1}{\epsilon}(ln{(|H|)} + ln{(\frac{1}{\delta})})\]](https://data-science-blog.com/de/wp-content/ql-cache/quicklatex.com-901f897bb0040e542c0eeaa14098b677_l3.png "Rendered by QuickLaTeX.com")
 Mrd. Punkten. Mit einer einfachen bool’schen Kodierung ergibt sich
Mrd. Punkten. Mit einer einfachen bool’schen Kodierung ergibt sich  und
und  .
. – wie auch immer – klassifizieren, so würden wir für den Einsatz von Naive Bayes oder unbegrenzten DecisionTrees mindestens 76.145 Datensätze benötigen. Weder die monatlichen Daten von Produkt A noch Produkt B würden ausreichen.
– wie auch immer – klassifizieren, so würden wir für den Einsatz von Naive Bayes oder unbegrenzten DecisionTrees mindestens 76.145 Datensätze benötigen. Weder die monatlichen Daten von Produkt A noch Produkt B würden ausreichen. kann etwas außerhalb des Hypothesenraums liegen, der durch das eingesetzte Lernverfahren erfasst wird. Dies bedeutet, dass wir im Hypothesenraum des Lernverfahrens nur eine Näherung
kann etwas außerhalb des Hypothesenraums liegen, der durch das eingesetzte Lernverfahren erfasst wird. Dies bedeutet, dass wir im Hypothesenraum des Lernverfahrens nur eine Näherung  erlernen können, die möglichst gut sein sollte. Solch ein – als agnostisch bezeichnetes – Lernverfahren muss daher bestrebt sein den Fehler zwischen den Trainingsdaten und dem Fehler der sich durch das Erlernen der Näherung
erlernen können, die möglichst gut sein sollte. Solch ein – als agnostisch bezeichnetes – Lernverfahren muss daher bestrebt sein den Fehler zwischen den Trainingsdaten und dem Fehler der sich durch das Erlernen der Näherung ![\[m \geq \frac{1}{2\epsilon^2}(ln{(|H|)} + ln{(\frac{2}{\delta})})\]](https://data-science-blog.com/de/wp-content/ql-cache/quicklatex.com-0630d9d4a9a075de904ef5d5610c93eb_l3.png "Rendered by QuickLaTeX.com")




 . Dieser wird als Bias, selten auch als Default-Wert, bezeichnet. Der Bias ist also der Wert, wenn die
. Dieser wird als Bias, selten auch als Default-Wert, bezeichnet. Der Bias ist also der Wert, wenn die  ein Fehler
ein Fehler  existiert. Diesen Fehler wollen wir in diesem Artikel ignorieren.
existiert. Diesen Fehler wollen wir in diesem Artikel ignorieren. statt
statt  ) sind nichts anderes als Gewichtungen zwischen den Eingaben.
) sind nichts anderes als Gewichtungen zwischen den Eingaben.![\[y = w_{0} \cdot x_{0} + w_{1} \cdot x_{1} + \ldots + w_{n} \cdot x_{n}\]](https://data-science-blog.com/de/wp-content/ql-cache/quicklatex.com-19728ae5f9e3065a4c17d50c417e0a8b_l3.png "Rendered by QuickLaTeX.com")
 . Verkürzt ausgedrückt:
. Verkürzt ausgedrückt:![\[y = \sum_{i=0}^n w_{i} \cdot x_{i}\]](https://data-science-blog.com/de/wp-content/ql-cache/quicklatex.com-3bbcbb30d482e7a1fd8da528030d6561_l3.png "Rendered by QuickLaTeX.com")
![\[y = w^T \cdot x\]](https://data-science-blog.com/de/wp-content/ql-cache/quicklatex.com-939f4bae6617db519d13b502d6337ee1_l3.png "Rendered by QuickLaTeX.com")
 .
.
 . Auf der linken Seite finden wir alle Eingabewerte, wobei der erste Wert statisch mit 1.0 belegt ist, nur für den Zweck, den Bias (
. Auf der linken Seite finden wir alle Eingabewerte, wobei der erste Wert statisch mit 1.0 belegt ist, nur für den Zweck, den Bias (
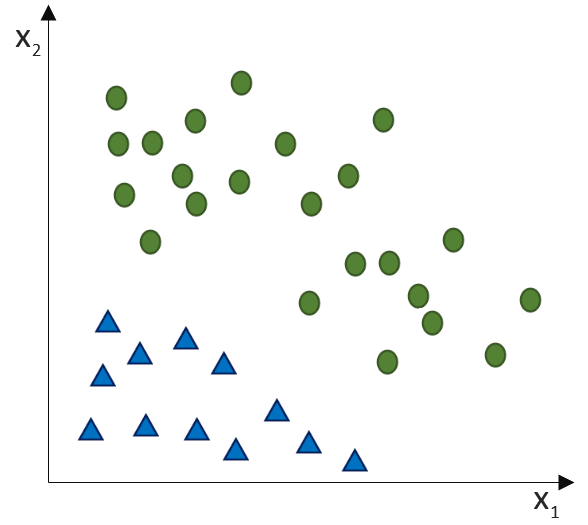
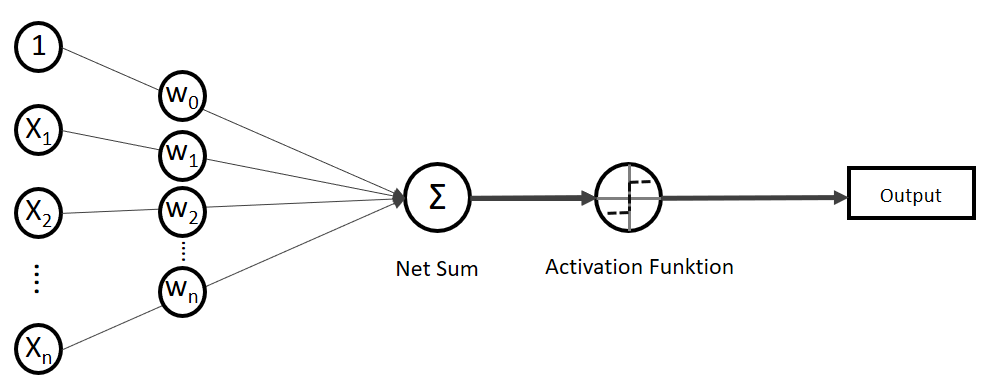
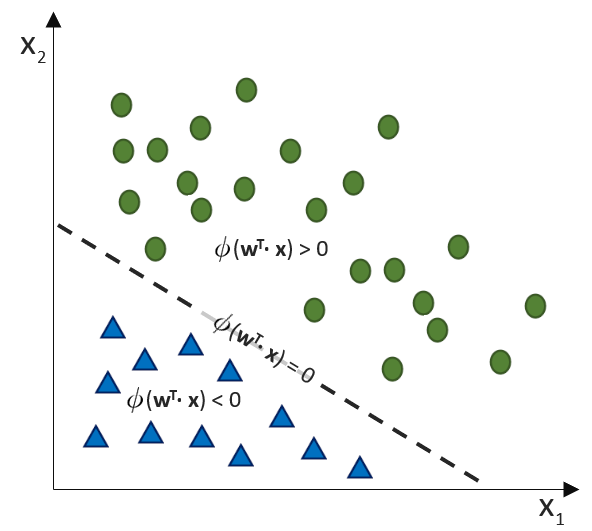
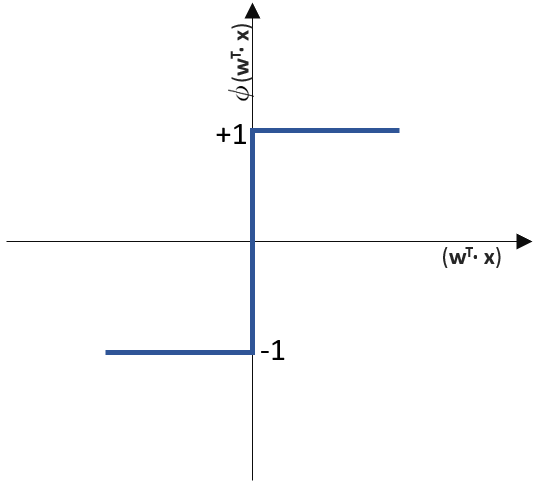
![\[ y = \phi(w^T \cdot x) = \left\{ \begin{array}{12} 1 & w^T \cdot x > 0\\ -1 & \text{otherwise} \end{array} \]](https://data-science-blog.com/de/wp-content/ql-cache/quicklatex.com-7b828cf4bbabf9e1a84ec9b628d51249_l3.png "Rendered by QuickLaTeX.com")
![3D Scatter Plot in Python [Matplotlib]](https://data-science-blog.com/de/wp-content/uploads/sites/5/2016/04/3d-scatter-plot-immobilien-klassifikation-gap.jpg)